
晶体化合物
吡咯中氮气中的低电子密度使它们成为弱碱(对于吡咯,Kb约为10?17;cf约为10?5),不可能用水酸制造吡咯盐。
事实上,正如接下来所显示的,质子化发生在碳上,而不是在氮上。
此外,吡咯不会与烷基化剂形成季盐,也不会与过氧基化合物形成胺类氧化物。
这与吡啶形成了鲜明的对比。吡咯中氮上电子对不可用的另一种解释是,如果芳香六分体(及其稳定能)用于形成键,就会被破坏。
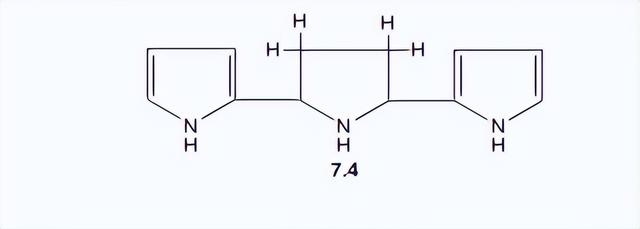
当吡咯被强酸加热时,会形成一个包含三个吡咯单元的晶体化合物。
其结构已确定为7.4。强酸也会导致吡咯产生不良的聚合物产物。这些过程依赖于环上碳的质子化,而不是氮的。

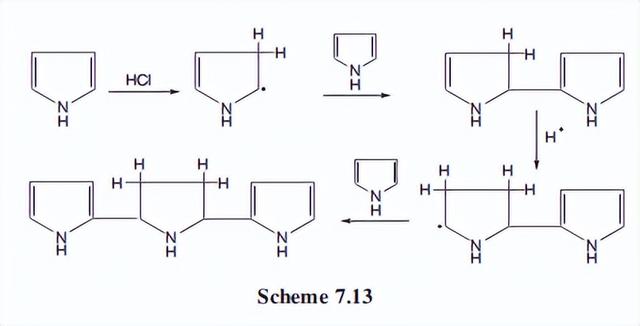
为了说明这个产品,我们提出了方案7.13的作用机制。这里,一个吡咯分子在3位质子化;这种情况并不常见,但并不未知。
由此产生的碳正离子然后作为一个亲电试剂指向第二个吡咯分子,该分子攻击通常的2位。中间体在二氢吡咯环的3位被质子化,形成的碳正离子攻击第三个吡咯分子的2位。
在设计合成物时,必须考虑到吡咯对酸的敏感性,并且在一般情况下,要避免使用酸性条件。
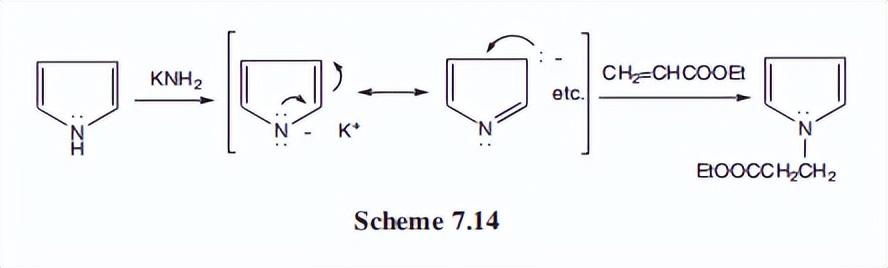
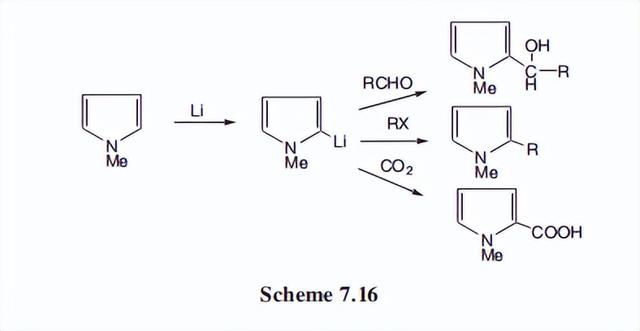
众所周知,仲胺的氢是弱酸性的,可以用强碱去除,形成阴离子。吡咯在氮上的氢酸性要大得多,可以很容易地用金属酰胺(如KNH2)等碱和活性金属(Na或K)去除。
吡咯的酸解离常数为10?17,与酒精的含量差不多。
吡咯阴离子的额外稳定性来自于负电荷的共振离域化。
吡咯阴离子可以被烷基化或酰基化,并可以参与迈克尔反应和其他典型的氮阴离子反应。
这些性质在n-取代吡咯的合成中非常有用。迈克尔反应如下图所示。
迪尔斯-阿尔德反应
苯和吡啶的芳香环体系阻止这些化合物以二烯的形式进入二烯-alder环加成反应。吡咯也是如此。
然而,通过在氮上放置吸电子基团,可以调节电子在环内的离域,因为这些基团共同接受n上的电子对。
例如吡咯的N-碳氧基衍生物;具有强亲二烯体,Diels-Alder环加成,得到7-azabicyclo七二烯(7-氮硼二烯)体系的衍生物。
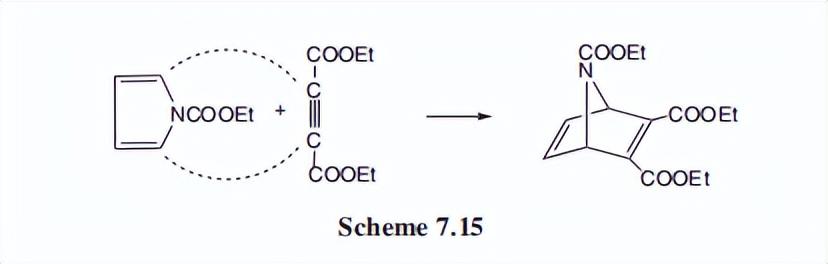
该方法对该桥接环体系的构建是非常有用的。
吡咯2位的金属化
- 烷基吡咯的一个综合有用的性质是,2位上的质子可以被活性金属(Na、Li和K)或强碱(丁基锂和BuLi)去除。
- 这种行为不同于缺氮杂环,在缺氮杂环中,强亲核试剂如BuLi添加到环C=N单元。
吡咯阴离子对亲电试剂反应,产生含有碳基的加成产物,或含有烷基的C-烷基衍生物。
呋喃的反应
呋喃是N、O、S杂环中芳香族最少的一种,在亲电取代反应中需要特殊的条件来保存环。这个环对酸性条件也很敏感。
当替换成功时,输入的是2个位置,其原因与吡咯替换的描述相同。一些对呋喃有用的工艺描述如下:
磺化。吡啶中的三氧化硫得到2-磺酸
断言。二恶烷中的溴得到2-溴衍生物。不需要催化剂。
硝化。预成型的氟硼酸硝基铵(二氧化氮+氟硼酸盐?)作用于生成2-硝基衍生物。
酰化。与乙酸酐反应得到2-乙酰基衍生物。
在某些条件下,呋喃在二烯体系中加入反应物,从而得到非芳香族产物。这是通过与亲电试剂反应形成通常的碳正离子而发生的。
然后添加亲核试剂而不是以通常的芳香取代方式消除质子。例如,当使用甲醇溶液中的硝酸作为硝化介质时,该产物为5-甲氧基-2-硝基加成产物。
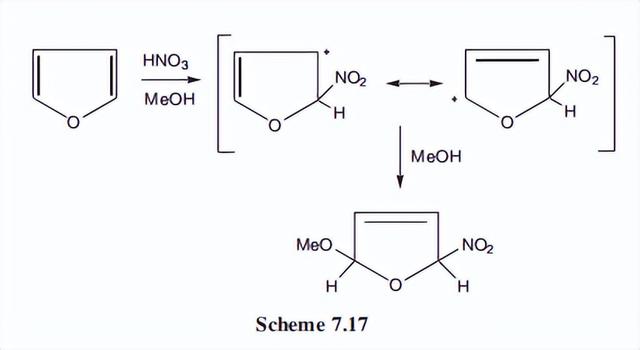
另一个例子是在水中进行溴化反应,在这里,溴化离子作为亲核试剂。

与其他芳香体系相比,呋喃在Diels-Alder反应中表现良好,许多例子是已知的。与马来酸酐的反应见方案。
在这里,内显加成产物的形成速度比exo快,但外显异构体更稳定。
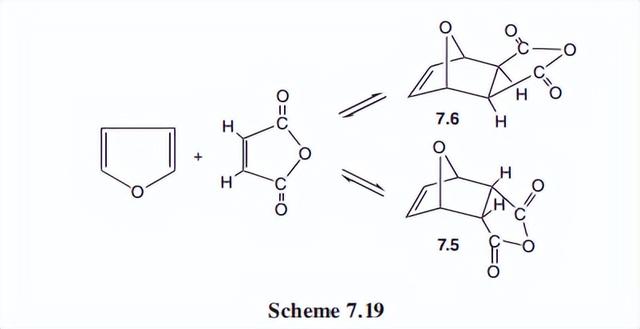
因为这是一个平衡的过程,最终的exo是最终的产物。我们已经证明了呋喃是分子内Diels-Alder反应的优秀参与者。
噻吩反应性
噻吩经历了大量的亲电取代,有些比苯更容易,这可以从环中pi-电子密度的增加中预期出来。替换发生在2个位置。
下面列出了一些对噻吩取代有用的特殊条件的例子。
磺化作用在30?C下发生在浓硫酸中。
硝化是用硝酸-乙酸酐完成的。
在黑暗中氯化,或与硫酰氯一起氯化,得到单氯衍生物。
与乙酰氯的弗里德尔工艺酰化需要较温和的催化剂四氯化锡,而不是通常的氯化铝。
用Me2NCHO和三氯氧磷进行甲酰化反应,可以很容易地将醛基安装在环上。
磷孔环系统
原则上,PH取代NH的环体系应该具有芳香特性,因为P上的孤电子对可以像吡咯那样被离域到环中。
然而,母磷孔分子是不稳定的,仅在?90?C,11的光谱上检测到,但p-甲基衍生物是稳定的和可蒸馏的,还有许多其他已知的取代磷孔。
该领域正在定期发展,值得在这里简要考虑。这些文献已经得到了广泛的综述。
关于磷孔具有芳香性质的现实已经有了很多讨论。第二排元素参与离域现象的一般观点没有错;毕竟,周期体系中磷的邻居是硫,噻吩是一种高度芳香的化合物。
磷的问题是,与氮完全不同的是,当这个原子处于三价态时,这个原子具有sp3杂化和稳定的锥体形状。
这意味着具有三个不同基团的磷是手性的,而且具有光学活性的磷烃确实是已知的。金字塔反转的活化能约为30-35千卡/摩尔,足以使简单的膦在回流二甲苯中的半衰期约为3小时。
14在室温下,孤对定位于sp3轨道,而不是p轨道,因为吡咯中的势垒较低。sp3轨道朝向远离环的平面,因此不能像与氮所发生的轨道那样有效地与碳孔隙重叠。
人们普遍认为磷酸孔具有低芳香性。然而,需要注意的是,质子核磁共振谱确实显示了芳香体系中发现的前场移动。
对于1-甲基磷孔,在复杂的AABBX谱(X = 31P)中,质子和质子都在δ为6.5-7.5。简单的磷孔通常是液体,但1-苄基磷孔在室温下被发现是结晶固体。
这使得该结构可以通过x射线衍射分析来确定,这清楚地显示了磷的锥体形状。
然而,从环内键长的测定表明,存在一些电子离域;这给了较低的BI为35.5、2、7、16,低于呋喃(43),但仍然指出了离域的测量。
NICS值也远低于呋喃的NICS值。这是通过将大的2,4,6-三(叔丁基)苯基取代基放置在P环上的实验完成的。
x射线衍射分析确实显示P金字塔明显扁平,环内键长有明显的变化。17鸟类指数为56.5,接近吡咯的值(59)。
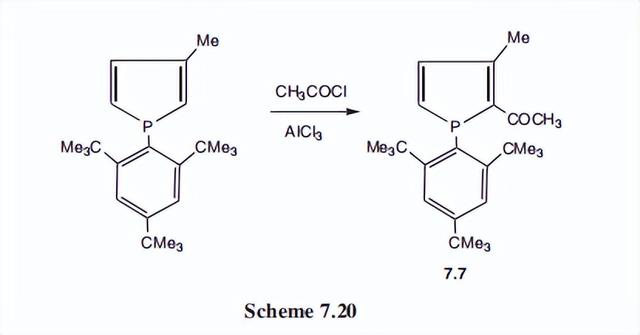
与这种增加的芳香性相一致的是,该化合物与乙酰氯和氯化铝进行了弗里德尔-克拉夫特反应,这是第一个在碳上发生任何亲电取代的磷酸孔。
主要产物的结构为7.7;酰化作用也发生在第4位和第5位。磷孔的其他性质是磷所特有的,而且大部分与吡咯的性质不同。
吡咯的苯并衍生物(铟)
海豚科是所有杂环科中最重要的一种,而且该系统的化学性质是巨大的。许多天然产物和合成药物都含有这个细胞核。
我们在这里局限于芳香性问题,我们发现我们正在处理的是一个稳定的环系统,它展示了过量所预期的所有特性。计算出的电子密度如图下图所示,其中发现所有碳的密度都大于1.00。
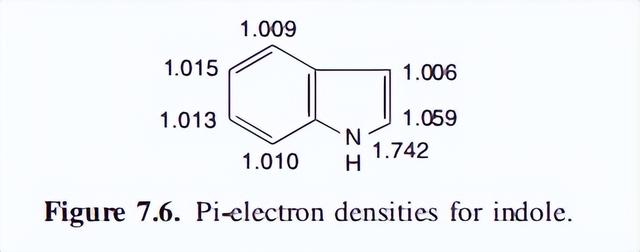
对3位的偏好可以通过考虑一个正物种攻击的共振可能性来解释。中间体7.8通过氮的电子释放来有效地稳定,而正中间体在2位攻击时的稳定则需要破坏苯的芳香体系。
虽然它仍然是一种稳定作用,但不如7.8中直接电子释放。
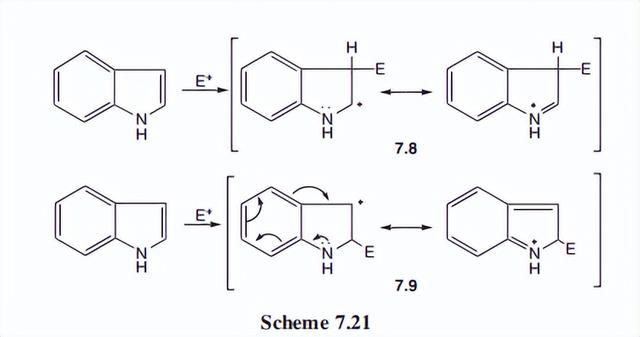
取代剂的试剂和温和条件类似于吡咯。应该补充的是,质子化发生在3位而不是氮气上,这并不奇怪。
大环Pi-过量的杂环
利用包含8个双键电子和氮上孤对的9元偶氮环体系,可以建立具有10个电子(n = 2)的高温芳香体系。用一个反式双键,应变被解除,化合物7.11具有合理的稳定性。

7元氮卓环的衍生物是众所周知的,尽管该环没有具有8个pi-电子的芳香族。事实上,根据它对未填充键合MOs的描述,它被归类为反芳香族。
大环氧和硫杂环也已知,具有4n+2pi-电子的杂环表现为芳香性。
具有混合杂原子结合的芳香族环体系
在这里,我们将考虑在一个5元环中有一个饱和的杂原子(例如,NH,O,或S)作为一个电子供体,以及一个类似吡啶的C=N作为受体的情况。
这种情况导致了强烈的离域化和具有巨大的稳定性和常见的环
咪唑系统为考虑这种情况提供了一个合适的例子。然而,在NH基团中,存在互变异构体,质子从一个氮转移到另一个氮。
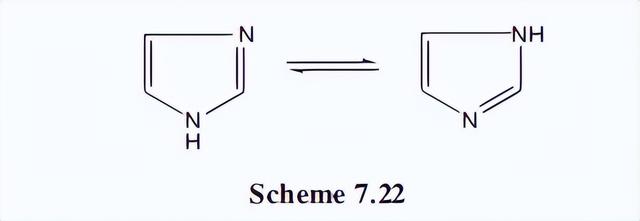
为了解决这个问题,我们将考虑咪唑的n-甲基衍生物,其中不可能存在互变异构体。该化合物的共振杂化反应如下图所示。
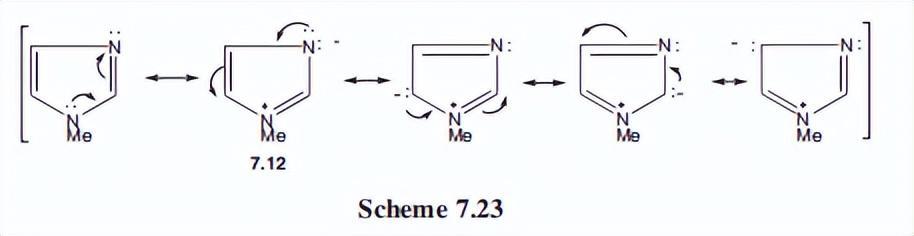
作者认为:贡献者说明了供体N-Me和受体C=N的直接相互作用,这是杂种的主要贡献者。N上的负电荷是可以接受的,当然也与吡啶型离域化有关。
结果表明,咪唑的IA为79,与吡咯(85)和噻吩(81.5)的IA比较接近。
1,3-恶唑中的氧电子和1,3-噻唑中的硫电子的共振杂化是相同的形式。
在噻唑中,鸟的IA值为79。这些化合物具有芳香族体系的所有特征,并且容易进行亲电取代。
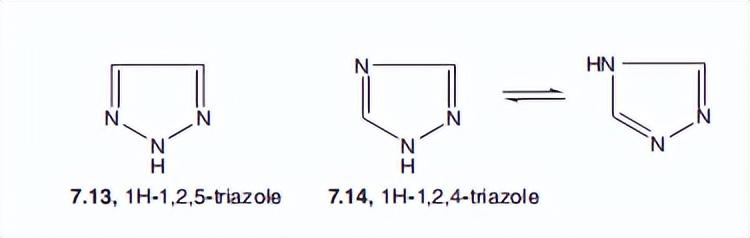
同样,在三唑类药物7.13和7.14中也存在明显的离域现象。鸟的IA值甚至更高(分别为90和100)。
来自杂原子O、N和S的不同混合物的相当多的杂环体系是可能的,并且在存在多环受体的情况时,芳香性可以预期是明显的。
这些原理也适用于双环和多环体系,并且已知大量稳定的芳香族体系。嘌呤是最重要的双环体系之一,它存在于核酸的碱基中。
在这里,一个嘧啶环和一个咪唑环被融合。后者将表现出其通常的互变异构行为,但现在这两种形式并不相等,而嘌呤本身的7.15形式已知在7.16中占主导地位。
参考文献
- 溴代吡咯腈高效液相色谱分析[J]. 吴红,焦九英,蒋松钰,李晓燕. 科技信息. 2013(09)
- [2]2-(溴代吡咯腈-1-基)乙羧衍生物的设计、合成及生物活性[J]. 周蒲,张列雄,马俊豪,郭倩男,游江,昌勤哲,徐志红. 农药学学报.
- [3]溴代肟醚丙酮溶液的气相色谱法分析[J]. 王立娜,杨娇,闵怡,梁文帅. 化工设计通讯. 2021(11)
创业项目群,学习操作 18个小项目,添加 微信:jjs406 备注:小项目!
如若转载,请注明出处:https://www.xmfxquan.com/12822.html
相关推荐
-
沈阳小伙儿狂喷三十分,沈阳小伙狂喷30分词.
接上篇: 自从肖军把我追到手,他再也不去仁和饭店了,以前,他就像上班似的准时准点,早上开门就去,晚上饭店打烊他回家,一辆破旧的自行车,拉着他风雨无阻,不是去饭店,就是在去饭店的路上…
-
鸡胸肉七种吃法,鸡胸肉的12种经典做法?
一、鸡胸肉的七种吃法 1、水煮鸡胸肉:将鸡胸肉切成片状,放入开水中煮熟,捞出晾凉后配上蒜泥或者姜丝调味即可。 2、鸡胸肉沙拉:将鸡胸肉切成丝状或块状,再加上蔬菜沙拉、低脂酸奶、各种…
-
谷雨,谷雨护肤品?
谷雨护肤品——秋季皮肤健康守护神 一、谷雨的意义 谷雨,是中国二十四节气之一,表示季节的变化,也是农民朋友们播种下一季作物的开始。在这个节气,气温升高,空气湿度增大,人的代谢能力增…
-
调职申请书(调职申请书格式范文)
一、调职申请书的目的 调职申请书的目的是请求公司的领导批准将自己从目前的职位转移到其他职位来适应自身的发展需要和公司的需要。 在申请调职时,应当明确表达出想要调职的原因、调职后的工…
-
母亲节送什么花比较好,母亲节送什么花比较好一点?
一、母亲节送什么花比较好 母亲节送花是表达对妈妈无尽的爱的一种方式,但是该送哪种花呢?最好选择一些寓意深刻的花,例如康乃馨、粉色玫瑰和百合等。 康乃馨是母亲节最受欢迎的花之一,它象…
-
肇东网络营销(肇东网络营销招聘)
肇东网络营销的魅力与招聘需求 随着数字化时代的到来,网络营销在各行各业中扮演着至关重要的角色。而肇东网络营销作为行业的先驱之一,吸引了无数人才的眼球。本文将探讨肇东网络营销的魅力以…
-
凤城网络营销(凤城网络营销招聘)
近年来,随着互联网的迅猛发展,网络营销行业也日趋兴旺,成为了众多企业推广产品和服务的首选方式。凤城网络营销作为行业内的领军企业,不仅拥有丰富的经验和技术,更是为广大求职者提供了一个…
-
网上做电商怎么做,电商怎么注册开店?
干货满满:要做电商,基础的逻辑还不去了解,先学基础,再去入行打狙击。因为这个行业很少人能自学成才,那么你就避免不了。 如果你没有基础认知,你会问一些比较离谱的问题,比如炒菜为什么要…
-
关于爱的故事电影赵平是谁演的,关于爱的故事电影赵平是谁演的啊?
2016年2月16日清晨,浙江宁波警方接到群众报警电话,说慈溪城小区门口发现了一具女性尸体。 接到报警电话后,警方立马出警到达案发现场,经勘察,死者胸部腹部身中多刀,随后经调查,死…
-
北京吃的特色美食有哪些,北京吃的特色美食有哪些?
一、 炸酱面 炸酱面是北京最有名的小吃之一,是一种传统的面食。它由白面条、豆瓣酱、酱肉末、黄瓜丝和葱花等佐料组成。其主要特色是豆瓣酱的浓郁和猪肉酱的香甜。最流行的炸酱面是来自老字号…