
文 | 青橘科普
编辑 | 青橘科普
前言
芳香化概念在有机化学中有着悠久的历史,回顾任何有机化学教科书中的讨论,我们将从这些讨论中挑选一些主要的思想和术语,以便它们可以应用于杂环。
我们首先需要认识到,芳香性不是一个明确的物理性质。
人们使用了各种策略来衡量它对其他特性的影响,并多次尝试给它加上数字。
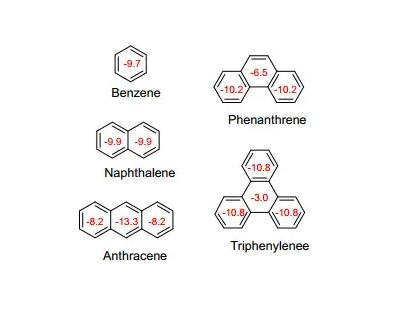

为了我们的目的,我们可以从三个理论的角度来考虑芳香性的描述,所有这些观点都解决了苯的pi-电子是离域的,而完全不像孤立双键中的pi-电子。
共振视图
共振理论是在1930-1940年期间发展起来的,虽然它很古老,但在有机化学中仍然是一个有用的工具,在杂环化学中尤其如此。
我们将广泛地使用它来解释反应机制,光谱效应,并在这里的芳香性的研究。
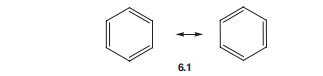
在共振理论中,苯的电子结构可以用两个Kekul‘e公式表示,如在共振杂化6.1中,两者都不是真实的。
但电子是离域的,并由分子整体共享。
因此,苯中没有单键或双键,所有键的实验长度均为相同的1.40?A,介于sp2-sp2单键(1.46?A)和双键(1.34?A)的值之间。该环是平面的,所有内键角均为六边角,长度为120?。
共振(或离域)的普遍存在,是一种能量还原效应,这在苯的情况下是明显的。
通过对燃烧热或加氢反应热的实验测量发现,具有三个真正双键的分子的能量比预期的少36千卡/摩尔,这个能量值通常被称为共振能量。
但我们需要记住,这是一个系统所没有的能量,而不是它所拥有的能量。
轨道视图
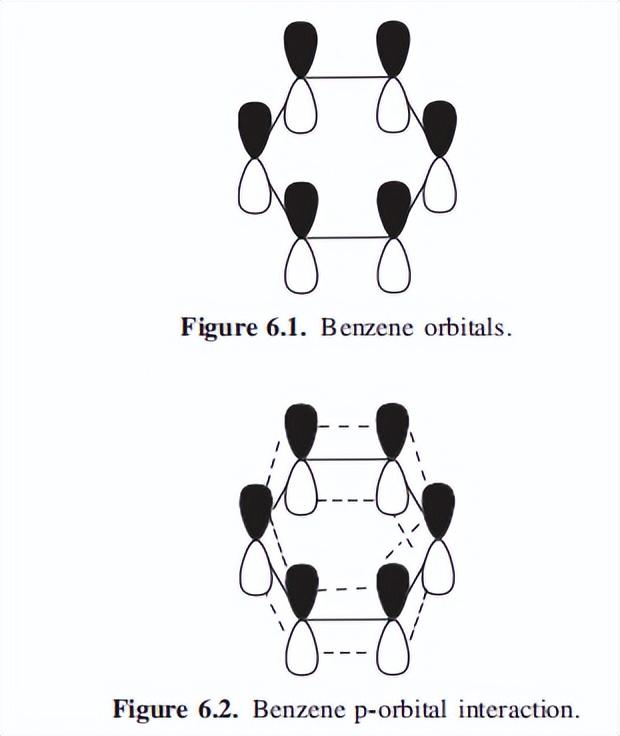
碳在苯中的杂化反应,为sp2(与120?的六角形中的键角一致)。
在每一个碳上,这将留下一个电子(称为pi电子)在环的平面上和下面的p轨道上,这一点如图6.1所示。
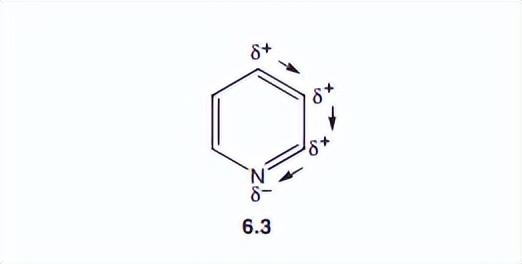
p轨道的重叠会在环的上方和下方,形成一个均匀的微电子密度区域。重叠的轨道如图6.2所示,这导致了一个众所周知的苯的视图,对环平面上方和下方的重叠轨道,有一个类似甜甜圈的描述(图6.3)。
轨道视图有助于理解苯在金属配位中,作为pi供体以及在与亲电试剂反应中,作为亲核中心的功能。
分子轨道视图
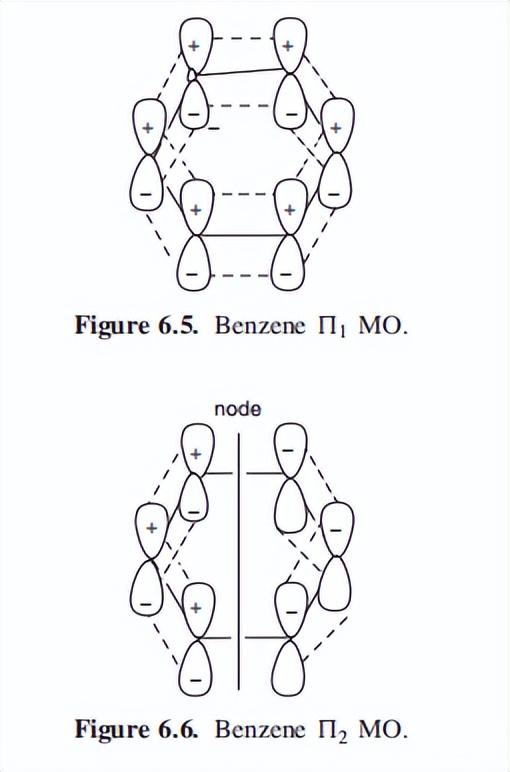
根据第五章简要回顾的分子轨道(MO)理论,苯的六个原子轨道将形成六个分子轨道。
有三个粘接MOs和三个反粘接MOs,它们的相对能量和电子分布如图6.4所示。
注意,在最高占据的Mo(HOMO)(2)中,有两个能量相等的填充轨道,这些轨道据说是“简并的”。
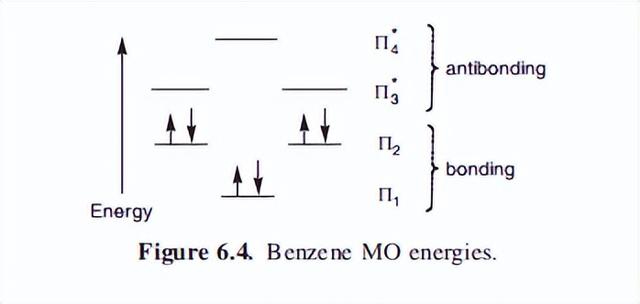
从Mo理论可以得到一个新的芳香性定义:在一个循环的完全不饱和系统中,如果没有未填充的键分子轨道,则系统是芳香的,但如果这些MOs只是部分填充,则系统是反芳香的。
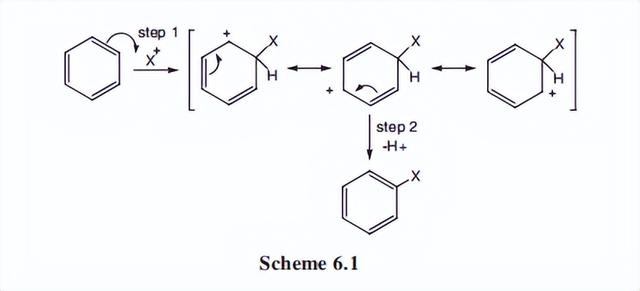
亲电取代的机理
苯和其他芳香烃在被亲电试剂攻击时的行为,不同于烯烃。
烯烃添加亲电物质,芳烃通过亲电中心取代环质子。取代H+的常见物种有X+(来自氯气或溴)、二氧化氮+(来自硝酸)、R+(来自R-X)、SO3、R(O)C+(来自RCOX)等。
与这些物种的反应也发生在芳香杂环化学,这将被讨论。
以X+作为亲电试剂为例,根据方案6.1的机理,可以分两步表示取代反应。共振理论在这里解释由亲电试剂附着形成的,中间体上的电荷能量减少扩散时是有用的。
核磁共振(NMR)的光谱特性
核磁共振波谱在芳香族和杂环化合物的研究中,具有巨大的价值,为了理解这本书中的介绍,建议读者在任何有机教科书中回顾这一主题。
在这里,我们将使用一些想法和术语,但我们不能深入到核磁共振的基础知识。
质子通常在参考四甲基硅烷(TMS,Me4Si)的,前场方向的信号为0 ppm。
脱硫是一种很小的效应,只有外加磁场的百万分之一(ppm),但仪器可以探测到它。正常的去除度为0-10ppm,但无一例外。在饱和化合物中,去抑制作用是由西格玛电子在外加磁场中的循环引起的,这就建立了一个二次磁场来加强外加磁场。
含碳上的电子密度,受1-3个键内的电子吸引取代基(大多数常见的杂原子)的影响,而与距离和角度相关的各向异性效应,主要来自双键和芳香环平面上下的pi-电子的循环。
pi-电子的影响比取代基的影响更强,双键上的质子通常在δ为5.0-5.5,而吸电子取代基通常引起烷烃C-H信号的前场偏移不超过3 ppm。
在附着在苯环碳上的质子上,我们发现电子循环产生的二次磁场发生了抑制,这个场增强了芳香C-H键区域的外加磁场,这样必须用来实现共振条件的场就减少了。
芳香族电子在应用场中循环时,被认为是在环周围形成电流。
这种所谓的“环电流”在建立二次磁场方面,比双键效应更有效,二次磁场也有一个区域在环的中心。
在环的上面和下面,这个区域正好相反,从而导致质子的屏蔽和这些位置的前场位移。
苯上的所有质子当然都是相同的,它的质子核磁共振谱由δ值为7.37处的一个单峰组成。
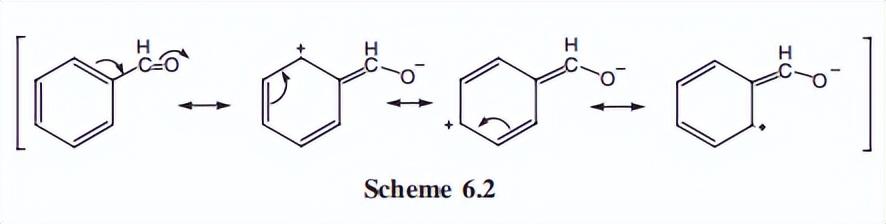
因此,很容易区分芳香族质子信号和烯烃的质子信号,环上的吸电子取代基降低了邻质子和对质子上的电子密度,构成了脱位效应。
因此,大多数这类化合物的光谱,具有大约δ 7.5到8.0的重叠信号复合物。
正质子和对质子更前场,元相对前场,这种差异可以很容易地通过,检查苯甲醛的共振结构来解释,部分正电荷只能出现在环上的邻位和对位。
就像1h一样,13C同位素的量子自旋数(I)为1/2,因此具有低能量自旋态和高能自旋态。
在核磁共振实验中得到了分辨率良好的峰,由于13C同位素的自然丰度较低(1.1%),光谱是通过傅里叶变换技术,通过多次扫描样品获得的。
位移通常由TMS测量,位移的范围超过200 ppm,几乎所有的信号都出现在TMS的前场。
饱和碳一般在δ范围为0-50,而sp2碳大约在δ范围为100-200。苯的碳在δ值为128.5,这些接近来自烯烃的信号,因此这里没有来自芳香环电流的特殊副作用。
环上的取代基经常引起较大的位移差异,主要是共振效应,前场吸电子,前场是电子释放。13C核磁共振对芳香族和杂环类化合物特别有用,我们将进行讨论。
1h和13C NMR之间的一个重要区别是,碳的积极性的增加,无论是通过共振还是诱导,都会使C处的p轨道收缩,这是一种抑制效应。
吡啶作为一个说明性模型的研究
吡啶是一种沸点相当低,为115?c的液体,它可以与水无限混溶,通过感应和共振效应证明了其极性特性。吡啶有一种强烈的气味,大多数人觉得这很令人讨厌。
吡啶中吸电子氮原子的存在对其性质有深远的影响,苯的共振图可以应用于杂化6.2中的吡啶。
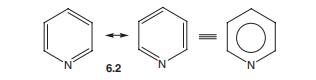
然而,电负性氮原子引起分子的显著极化,如结构6.3所示。
此外,吡啶可以显示额外的共振结构,其中连接双键的电子对可以放置在氮上(方案6.3)。
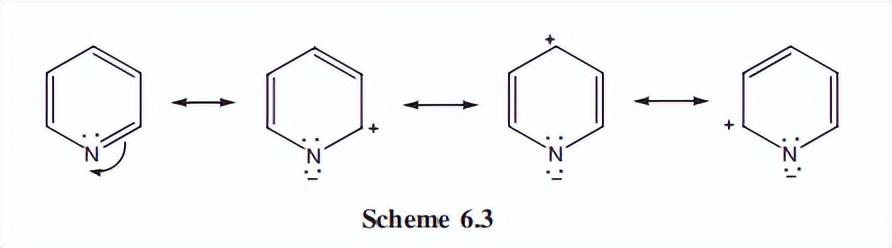
这种形式具有二价但带负电荷的N,这是N的稳定条件。
正电荷分散在环周围的碳上,特别是C-2和C-4(而不是C-3),净效应是降低了环中相对于苯的pi-电子密度,这是描述吡啶为pi-缺乏的来源。
苯在所有位置的pi-电子密度为1.0,计算结果如下:N1.166C-20.866C-31.064和C-40.932。这种效应适用于所有含有C-=-N单元的6元芳香环。
如图所示,5元环中的杂原子正好相反,将pi电子密度进入环,从而成为pi过度。
这些影响并非微不足道的,这两种环的性质有很大的区别。
实验发现吡啶的共振能在21-43千卡/摩尔范围内,与苯很相似。由于在杂环上进行传统的实验难以获得这个值,所以实验范围很大。
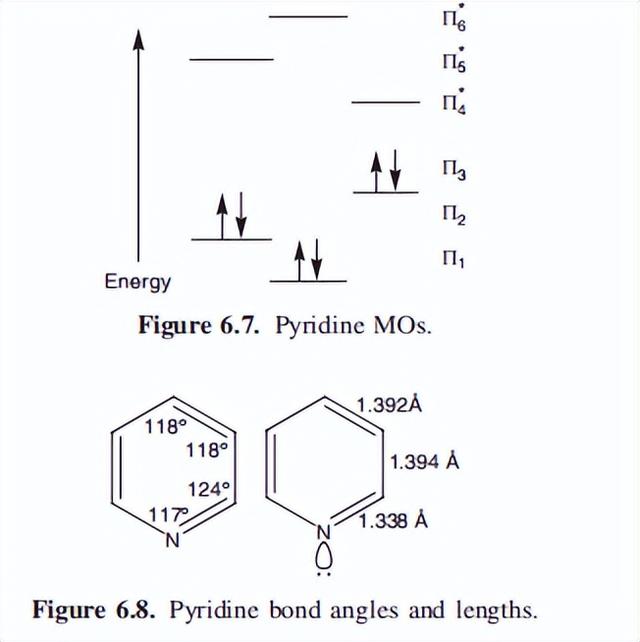
吡啶的分子轨道图如图6.7所示,这幅图和苯的图是有不同的。在苯中,HOMO由两个能量相等的填充轨道组成(因此是简并的),但在吡啶中,只有一个填充的HOMO。
重要的是,没有一个轨道是退化的。
电子诱导和共振离域化的联合作用,使吡啶成为一个具有相当极性的分子。
它的偶极矩为2.20 Debye (D)单位,这个值可以与哌啶(1.57 D)的值进行比较,哌啶的极性仅通过感应控制,吡啶作为一种非质子极性溶剂有许多用途。
作者观点
由于N是sp2杂化和平面的,整个吡啶分子是平面的,对于其他具有多个C=N单元的芳香族杂环也是如此。
苯的C到N键比苯的C到C键短,这导致环内的角度(图6.8),从观察到的苯的完美六角形的120?中被改变。
氮气上的孤对轨道落在环的平面上,这使得它可以与亲电键结合或与金属离子配位。
吡啶的特殊核磁共振特性就像苯一样,芳香环电流导致附着在环碳上的所有质子脱落。
然而,氮(结构6.3)的诱导效应导致电子从所有碳中退出,但随着它必须通过的键的数量的增加,它会减少,导致额外的分离,特别是在C-2,6附着在N上。
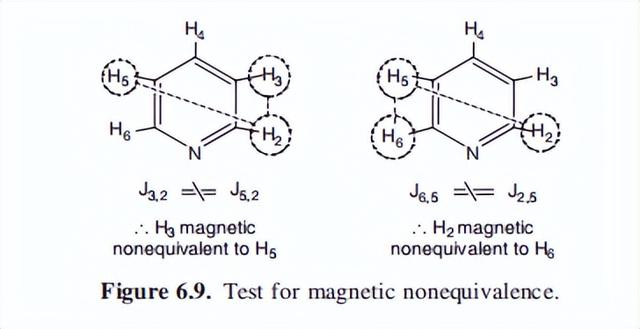
这就产生了一个复杂的频谱,在其中观察到许多信号分裂,一个简单的解释是不可能的,吡啶的光谱如图6.10所示,很明显,分裂比预期的一级光谱要多得多。
吡啶的光谱可以描述为AAMMX型,这一术语表明,核A、M和X的位移并不接近。
有两个核A是磁性不等价的,两个核M的磁性不等价性并不罕见,所有单取代苯、4取代吡啶、吡咯甚至一些非环化合物的质子核磁共振谱,都可以出于同样的原因表现出二阶谱。
参考文献:
苯并咪唑离子液体的合成及其催化尼泊金甲酯的合成[J]. 刘玉婷;邹倩;尹大伟.中国调味品,2021(03)
离子液体的性质和在萃取技术中的应用[J]. 孙思娜;刘书彤;陈庆阳;洪羽;黄涛;田鹏;段纪东.山东化工,2020(24)
创业项目群,学习操作 18个小项目,添加 微信:jjs406 备注:小项目!
如若转载,请注明出处:https://www.xmfxquan.com/12780.html
相关推荐
-
华蓥网络营销(华蓥网络营销招聘)
在今天竞争激烈的商业环境中,华蓥网络营销行业被认为是一条适合创业者的黄金之路。随着互联网的飞速发展,越来越多的企业开始关注网络营销的重要性,并投入大量资源在这个领域进行推广。然而,…
-
态生两厌之愁靥是指什么,态生两之愁中靥怎么念.
薛宝钗 宝钗黛玉二人如两峰对峙,互为映衬,互不相让,一静一动,一拙一巧,一个晶莹玉润,一个弱柳扶风。一个艳如太真,一个娇若西子,各具特色,各有风采。 第三十七回 秋爽斋偶结海棠社,…
-
淘宝利益点是什么意思小人之心度君子之腹!
大人看问题的角度和孩子看问题的角度不一样,也许我们认为的“对”在孩子看来可能是“错”。 女儿的QQ号被盗了,女儿的聊天交友工具成了犯罪份子的诈骗工具赚钱工具。 骗子通过QQ号,诈骗…
-
常见的网络营销工具有哪些(常见的网络营销工具有哪些_)
常见的网络营销工具有哪些? 随着互联网的广泛应用和快速发展,网络营销成为企业推广和宣传的重要手段。网络营销工具可以帮助企业更好地与目标受众进行互动,提高品牌知名度和销售效果。下面将…
-
常见的网络营销方式(常见的网络营销方式有哪些)
常见的网络营销方式有哪些? 随着互联网技术的飞速发展,网络营销已成为企业推广产品和服务的重要手段。在网络营销的世界中,有许多常见的方式能够帮助企业吸引目标群体的关注,提升品牌知名度…
-
打动人心的龙虾广告词怎么写,打动人心的龙虾广告词怎么写的?
薪侠客:龙虾烧烤。 想100个夏天的文案,都不及吃一场小龙虾。水漫桥南边的薪侠客,作为龙虾界的领头羊,次推出6个口味,先刷刷再炸炸。酥脆咸香的咸蛋黄口味,连壳都想给他嗦了。裹挟着虾…
-
网络营销书籍排行榜前十名(网络营销书籍排行榜前十名有哪些)
网络营销书籍排行榜前十名推荐 在当今数字化时代,网络营销已成为企业成功的关键。为了帮助广大营销从业者和创业者更好地了解和掌握网络营销策略与技巧,本文将为您推荐网络营销书籍排行榜前十…
-
幼儿园社会实践情况简单写,幼儿园社会实践情况简单写100字?
一、社会实践的概念 社会实践是指幼儿通过参与社会生活的各种实践活动,了解社会、认识社会、获得社会经验的过程。社会实践包括家庭实践、学校实践、社区实践等。 在幼儿园教育中,社会实践是…
-
网络营销的核心目标(网络营销的核心目标是)
网络营销的核心目标:突破边界,开创商机 随着互联网的快速发展和普及,网络营销已经成为现代商业中不可或缺的一部分。作为一种高效、便捷和广阔的营销手段,网络营销的核心目标是通过网络渠道…
-
《把悲伤留给自己》,《把悲伤留给自己》歌词.
家乡林则徐禁毒主题公园 又到了一年春节假期,很多亲朋好友都回家了,疫情三年来确实影响了彼此的联络,情感也放淡了不少,好在我们挺过来了,今年回老家大街小巷恢复不少人气,多了炮竹声响的…